Using machine learning to predict the potential risk of unknown chemicals
22 April 2025
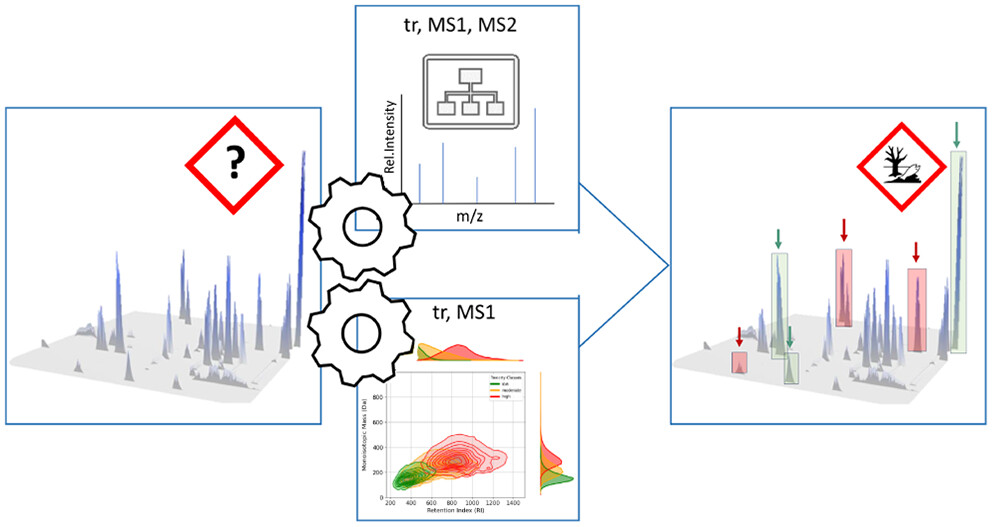
The study addresses a key challenge in environmental analysis: how to prioritize potentially harmful compounds without knowing their structures. Environmental samples often contain a wide variety of known and unknown substances, making their analysis time-consuming and challenging. Typically, less than 10% of the detected chemical signals can be directly connected to a single chemical structure.
To improve this, the researchers developed a data driven approach taking advantage of the chromatographic and mass spectrometric behaviour of unknown chemicals to predict their potential risk. They used a combination of two Machine Learning models to accurately predict the toxicity category of unknown chemicals measured by LC-HRMS. The approach marks a significant step toward more efficient and scalable environmental monitoring, helping to better understand and respond to ecological risks from unknown contaminants.
Abstract, as published with the paper
Complex environmental samples contain a diverse array of known and unknown constituents. While Liquid Chromatography coupled with High-Resolution Mass Spectrometry (LC-HRMS) Non-Targeted Analysis (NTA) has emerged as an essential tool for the comprehensive study of such samples, the identification of individual constituents remains a significant challenge, primarily due to the vast number of detected features in each sample. To address this, prioritization strategies are frequently employed to narrow the focus to the most relevant features for further analysis.
In this study, we developed a novel prioritization strategy that directly links fragmentation and chromatographic data to aquatic toxicity categories, bypassing the need for individual compound identification. Given that features are not always well-characterized through fragmentation, we created two models:
- a Random Forest Classification (RFC) model, which classifies fish toxicity categories based on MS1, retention, and fragmentation data—expressed as cumulative neutral losses (CNLs)—when fragmentation information is available, and
- a Kernel Density Estimation (KDE) model that relies solely on retention time and MS1 data when fragmentation is absent. Both models demonstrated accuracy comparable to structure-based prediction methods.
We further tested the models on a pesticide mixture in a tea extract measured by LC-HRMS, where the CNLs-based RFC model achieved 0.76 accuracy and the KDE model reached 0.61, showcasing their robust performance in real-world applications.
Paper details
Viktoriia Turkina, Jelle T. Gringhuis, Sanne Boot, Annemieke Petrignani, Garry Corthals, Antonia Praetorius, Jake W. O’Brien, and Saer Samanipour: Prioritization of unknown features based on predicted toxicity categories. Environ. Sci. Technol. 2025, published 20 April 2025. DOI: 10.1021/acs.est.4c13026
See also
Samanipour research group: Environmental Modeling & Computational Mass Spectrometry